Le Infezioni in Medicina, n. 3, 333-338, 2025
doi: 10.53854/liim-3303-11
CASE REPORTS
Late-Onset Combined Immunodeficiency presenting with Progressive Multifocal Leukoencephalopathy and associated Immune Reconstitution Inflammatory Syndrome
Luca Mezzadri1,2, Maria Lucia Borghesi1, Patrizia Comoli3, Maria Rosa Pozzi4, Giuseppe Lapadula1,2, Marianna Rossi1, Paolo Bonfanti1,2
1Infectious Diseases Unit, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy;
2School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy;
3Cell Factory and Center for Advanced Therapies and Pediatric Hematology/Oncology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy;
4Rheumatology Unit, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy.
Article received 25 April 2025 and accepted 8 July 2025
Corresponding author
Luca Mezzadri
E-mail: l.mezzadri3@campus.unimib.it
SummaRY
Progressive multifocal leukoencephalopathy (PML) can rarely occur in individuals with occult immunosuppression. Here, we describe the case of an adult man who presented with PML, in whom CD4+ lymphocytopenia and hypogammaglobulinemia were subsequently identified, leading to a diagnosis of late-onset combined immunodeficiency. Intravenous immunoglobulin replacement therapy was initiated. His clinical course, though complicated by immune reconstitution inflammatory syndrome, was favorable.
Keywords: Late-onset combined immunodeficiency (LOCID), progressive multifocal leukoencephalopathy (PML), Immune reconstitution inflammatory syndrome (IRIS), occult immunosuppression, opportunistic CNS infections.
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML) is a demyelinating neurological disorder caused by the reactivation of JC virus (JCPyV), which typically affects individuals with profound immunosuppression, such as those with acquired immune-deficiency syndrome (AIDS), organ transplant recipients, individuals with hematological malignancies, and those undergoing immunosuppressive therapy [1]. While PML is commonly associated with severe immunosuppression, it can occasionally occur in immunocompetent individuals or those with occult immunosuppression. The incidence, frequency, and prognosis of PML in individuals with previously unrecognized immunosuppression remain poorly understood and infrequently reported in the literature. However, a recent study suggests that these may account for over 10% of all reported cases [2].
In this report, we describe a challenging case of PML in a patient with occult immunosuppression, complicated by immune reconstitution inflammatory syndrome (PML-IRIS).
CASE PRESENTATION
A man in his early sixties presented at the emergency department of a Northern Italy hospital in the second half of 2022, exhibiting sudden-onset visual field disturbances, language difficulties and
memory impairments. His medical history was characterized by hypertension, dyslipidemia, and ischemic heart disease.
A brain computed tomography (CT) scan revealed a hypodense lesion located in the left posterior parietal region. The patient was initially admitted to the Neurology Unit to investigate possible cerebral ischemia.
Brain magnetic resonance imaging (MRI) scan demonstrated extensive white matter abnormalities in the left parieto-occipital region, characterized by hyperintensity in T2 sequences. A suspicion of PML was raised. A lumbar puncture was performed, revealing clear cerebrospinal fluid (CSF) with normal opening pressure. CSF analysis showed a cell count of 1/mm³, protein levels of 41 mg/dL, and glucose levels of 61 mg/dL. Presence of JCPyV DNA (1620 copies/mL) was detected. The identification of JCPyV DNA in the CSF, combined with pertinent clinical and radiological findings, established a definitive diagnosis of PML according to the criteria of the American Academy of Neurology/Neuroinfectious Disease Section [3].
In the early stages of hospitalization, the patient exhibited a progressive clinical deterioration
characterized by the onset of right-sided brachio-crural hemiparesis and aphasia.
Following the diagnosis of PML, the patient was transferred to our Infectious Diseases Unit for
further characterization of his condition.
Additional investigations were carried out to investigate potential underlying causes. Comprehensive laboratory evaluations, including fourth-generation HIV assay and polymerase-chain-reaction (PCR) analysis for HIV-RNA in blood, as well as screening for autoimmune and inflammatory conditions, yielded negative results. Serological tests for borreliosis, syphilis, and toxoplasmosis also returned negative results. Furthermore, Interferon-Gamma Release Assay (IGRA) for tuberculosis, serum cryptococcal antigen, and blood PCR assays for BK virus, HTLV-1, and EBV-DNA all returned negative results. Notably, the patient exhibited lymphocytopenia, particularly affecting CD3-CD4+ cells (CD3 532 cells/μL, reference range: 860-2607; CD4 122 cells/μL, reference range: 493-1666) and CD19 cells (11 cells/μL, reference range: 72-520), in addition to hypogammaglobulinemia (IgG 400 mg/dL, reference range: 700-1600; IgM 30 mg/dL, reference range: 40-230 mg/dL; IgA within normal range 70-400 mg/dL).
Cotrimoxazole was initiated for prophylaxis against opportunistic infections. Additional imaging studies, including PET/CT scan, revealed multiple infradiaphragmatic and supradiaphragmatic lymphadenopathies, suggesting a possible lymphoproliferative disorder. However, histological examination of a lymph node showed small lymphocytes with low proliferative activity (Ki-67 5%) and a mixed B/T phenotype. Bone marrow biopsy showed no evidence of lymphoproliferative disease. These findings, along with hematological consultation, ultimately allowed exclusion of a lymphoproliferative disorder.
An immunological assessment was also conducted, revealing a low but detectable frequency of circulating JCPyV-specific T cells.
Considered the patient’s immunological profile featuring hypogammaglobulinemia and lymphopenia, as well as his initial presentation with an opportunistic infection, a diagnosis of Late-Onset Combined Immunodeficiency (LOCID) was made.
Supplemental therapy with intravenous immunoglobulin (IVIG) was initiated. Over the subsequent two-month hospitalization period, during which physical therapy to promote motor function recovery was provided, the patient’s condition gradually improved, despite residual hypostenia as a notable symptom. A lumbar puncture was repeated before patient’s discharge, revealing that JCPyV was no longer detectable. The patient was discharged with follow-up visits at our outpatient clinic.
A brain MRI repeated a few days after discharge, shown in Figure 1, displayed a noticeable increase in the extent of signal abnormalities on T2-weighted sequences, involving the bilateral supratentorial white matter. Multiple areas of uneven enhancement were also observed, widely distributed with a patchy pattern. These findings were deemed to be consistent with a diagnosis of PML-IRIS. Consequently, steroid therapy was initiated with dexamethasone at approximately 0.1 mg/kg/die. Three months later, a follow-up brain MRI demonstrated significant resolution of the IRIS-related findings, with reduced T2 hyperintensities, decreased mass effect, and near-complete resolution of contrast enhancement. A further PET/CT scan did not show any lymphadenopathy.
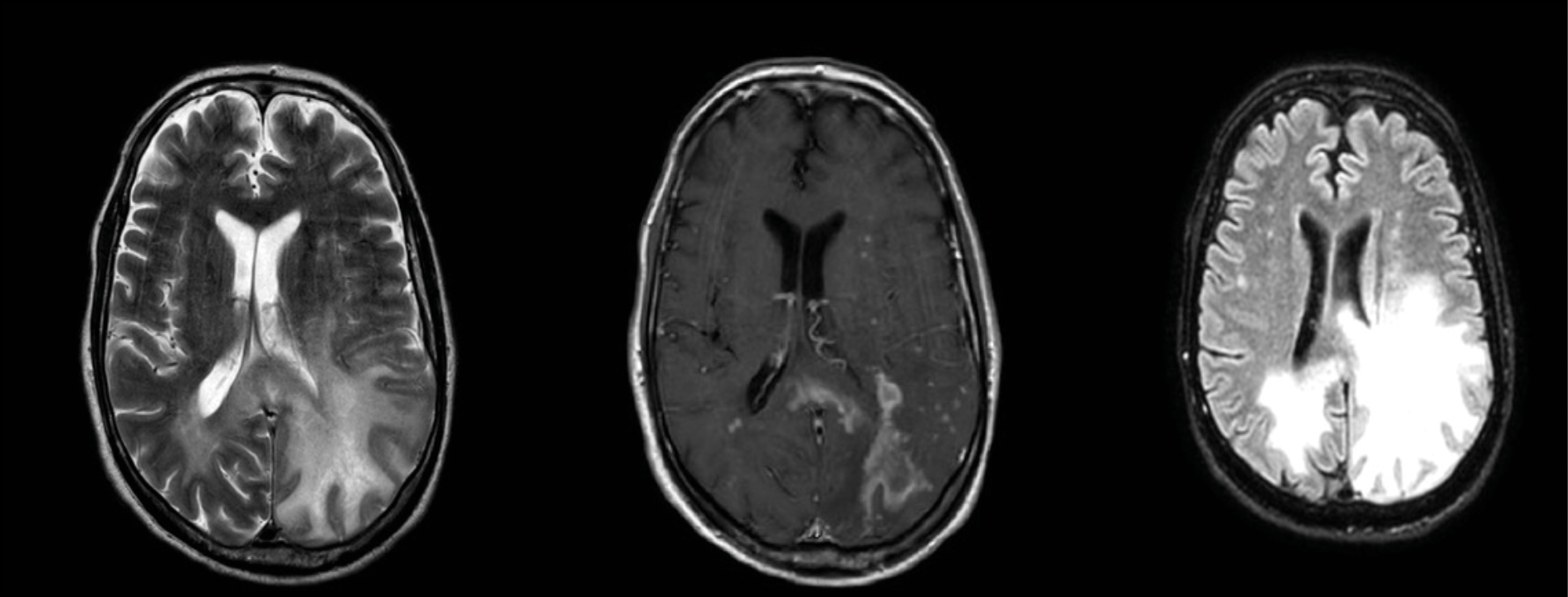
Figure 1 - Magnetic Resonance Imaging (MRI) scans illustrating a notable enlargement of hyperintense signal alterations in T2, primarily affecting the left supratentorial white matter with a mass effect indicative of interstitial edema. Multiple areas of patchy enhancement are concurrently highlighted.
Routine follow-ups at our outpatient clinic continued, including immunoglobulin infusions (0.5 g/kg every 4–6 weeks). The patient showed progressive cognitive and motor improvement.
These clinical events are summarized in the graphical timeline shown in Figure 2.
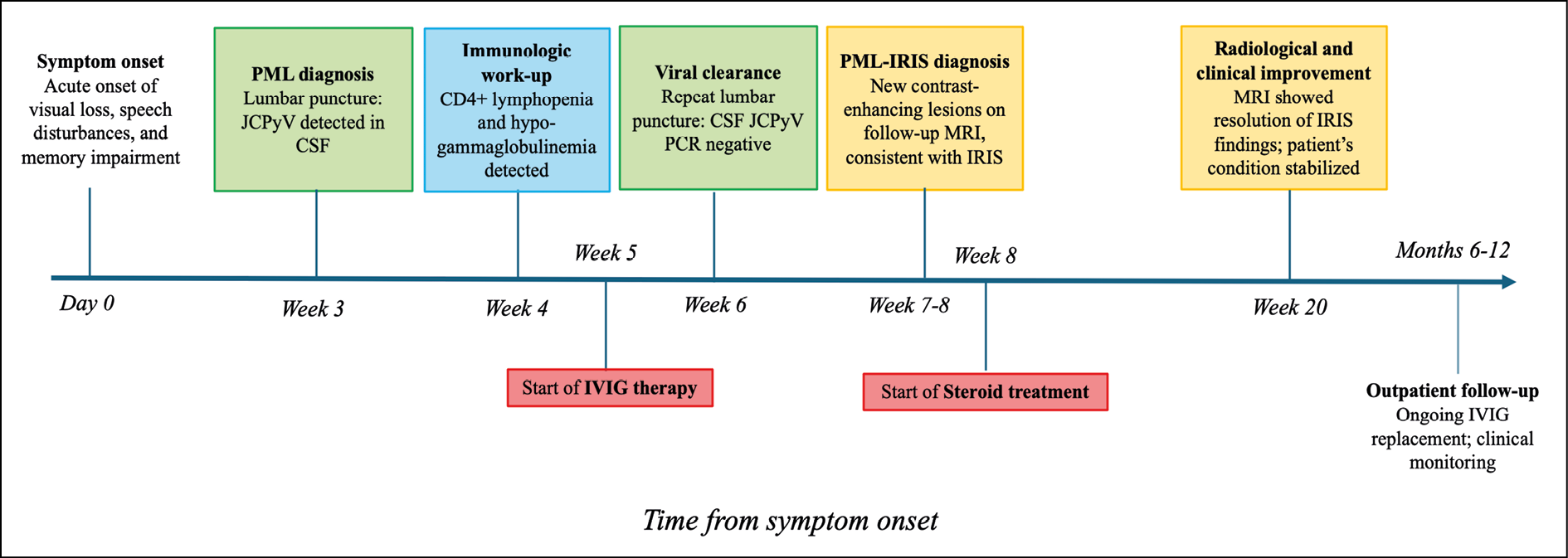
Figure 2 - Graphical overview of the patient’s clinical course from symptom onset through the first year of follow-up.
Abbreviations: PML: progressive multifocal leukoencephalopathy; JCPyV: John Cunningham polyomavirus; CSF: cerebrospinal fluid; IVIG: intravenous immunoglobulin; IRIS: immune reconstitution inflammatory syndrome; PCR: polymerase chain reaction.
Legend: Green boxes – Virological findings; Blue box – Immunologic findings; Yellow boxes – Radiological findings; Red boxes – Therapeutic interventions.
Approximately one year after the initial hospitalization, he was re-hospitalized at another institution due to the onset of seizures. However, repeat CT and MRI scans revealed no acute findings. A repeat lumbar puncture yielded negative results. Additional tests, including T-cell receptor rearrangement to rule out hematologic malignancies, were also negative.
Following discharge, the patient resumed follow-up at our outpatient clinic, where regular monitoring and IVIG therapy were continued. At our most recent visit, approximately 2.5 years after disease onset, the patient remains clinically stable. Mild aphasia and an unsteady gait persist, although significant improvement in brachio-crural hemiparesis was observed. Laboratory tests consistently show low CD4⁺ T-cell counts (<200 cells/μL) and IgG levels below the normal range. Consequently, IVIG administration has been maintained, along with ongoing prophylaxis with trimethoprim–sulfamethoxazole. No new neurological deterioration has been reported.
DISCUSSION
We presented a case of PML complicated by IRIS in a patient with latent immunosuppression, ultimately diagnosed with LOCID, a subset of common variable immunodeficiencies (CVID). LOCID, initially described by Malphettes et al. in 2009, is characterized by hypogammaglobulinemia, severe T cell deficiency, and late-onset opportunistic infections (OIs) [4]. Notably, PML was not reported among the OIs in the series by Malphettes et al. To our knowledge, this is the second case in the literature directly associating PML with a diagnosis of LOCID, following the report by Kurmann et al., although cases of PML in patients with features compatible with common variable immunodeficiencies have been previously documented [5]. Unlike the case reported by Kurmann et al., our patient had an unidentified immunodeficiency and was untreated, adding complexity to the understanding of the interplay between the syndromes.
JCPyV, a member of the polyomavirus genus, is the causative agent of PML. Acquired typically in early age and affecting 60-80% of the population by the age of 70, primary infection results in latency within specific cells, including precursor B cells, CD34+ hematopoietic progenitor cells, bone marrow, lymphoid tissue and tonsillar stromal cells. In addition, JCPyV DNA has also been detected in brain tissue of immunocompetent individuals as well as in immunocompromised patients without PML, suggesting that the Central Nervous System itself may serve as a site of latency [1].
Reactivation mechanisms, though not fully understood, suggest the crucial role of the T-cell immune system. Specifically, Du Pasquier et al. demonstrated that the early presence of JCPyV-specific CD8+ cytotoxic T-lymphocyte in peripheral blood is associated with PML containment [6]. In our case, the patient exhibited a weak immune response in the analysis, which may explain the slow progression of the clinical course. However, immunological testing was not repeated during follow-up, leaving the evolution of the immune response and its role in disease progression uncertain.
CD4+ T cells also play a significant role, with their counts correlating with PML prognosis [7]. Recent studies suggest also the involvement of the B cell compartment, as it was shown that human B cells secrete neutralizing antibodies against JCPyV and may serve as a viral reservoir [8]. Furthermore, according to current models of PML pathogenesis, prolonged impairment of cellular immunity may promote viral replication with subsequent emergence of neuropathogenic viral variants, particularly through rearrangements in the non-coding control region (NCCR) and mutations in the VP1 gene. Whether PML arises from reactivation of latent virus within the brain or from CNS entry of a peripherally reactivated, neuro-adapted virus remains unclear [1, 7, 9].
In this context, it is possible that the patient’s underlying immune dysfunction facilitated viral reactivation and neuro-invasion. However, JCPyV was not assessed in other compartments such as blood or urine, and sequencing of the CSF strain was not performed, limiting our further characterization of the infecting strain - an acknowledged limitation of our report.
HIV-negative patients with PML and immunodeficiencies typically face limited survival spans [10]. Various drugs, such as cytarabine, cidofovir, topotecan, mefloquine, and mirtazapine, have demonstrated in vitro activity against the JC virus. However, there is no evidence of in vivo efficacy [1]. While there is evidence suggesting the potential efficacy of infused JCV-specific T cells [11], this strategy was considered unnecessary in this case. Indeed, the patient exhibited a certain degree of immune response to the virus, that prompted a cautious monitoring approach to avoid onset of PML-IRIS.
The primary goal of treatment remains the restoration of underlying immunocompetence. In our case, initiation of IVIG therapy, the only treatment received by the patient, may have contributed to the favorable disease course. Immunoglobulin replacement represents a mainstay of treatment for primary immunodeficiencies involving defective antibody production, such as CVID, and is particularly effective in preventing recurrent bacterial infections [12]. In patients with LOCID, IVIG is administered at similar frequency to other forms of combined immunodeficiency, although the syndrome often presents with greater clinical severity, as evidenced by increased hospitalization rates despite comparable baseline immunoglobulin levels and replacement requirements [4]. Although robust evidence from large-scale studies is lacking, reports of successful use of IVIG in the treatment of opportunistic infections in patients with LOCID have been described [13, 14]. While its efficacy remains uncertain, IVIG replacement may act through both antibody replacement and immunomodulation, supporting CD4⁺ restoration, reducing CD8⁺ activation, and promoting immune homeostasis via effects on cytokines and dendritic cells [15, 16].
Other experimental strategies to enhance immune reconstitution in PML include immune checkpoint inhibitors and recombinant cytokines such as interleukin-7 (IL-7), which may support T cell expansion in lymphopenic patients by promoting naïve and central memory subsets [17-19]. However, a certain level of immune competence is required for these approaches to be successful, and, as many patients with PML have no virus-specific immune competence, benefits from these agents are difficult to anticipate. For these reasons, in this case their use was not attempted.
The occurrence of PML-IRIS in our case added significant complexity to clinical management. Immune reconstitution strategies are well known to trigger IRIS, particularly in patients with HIV initiating antiretroviral therapy who experience a rapid rise in CD4⁺ cell count or drop in HIV RNA, and in patients with multiple sclerosis following natalizumab discontinuation. Clinically, PML-IRIS may present as a paradoxical worsening of a known infection or as unmasking of a previously subclinical one [1]. In PML, the condition is typically driven by an exaggerated cytotoxic T-cell response that leads to marked CNS inflammation [20]. During immune recovery, JCV-specific CD4⁺ and CD8⁺ T cells emerge, particularly targeting the viral capsid protein VP1 [1]. Chemokine signaling, particularly involving the CCR5 receptor, has been implicated in the recruitment of these activated T cells into the CNS during this phase [21]. Histopathological findings show dense infiltration of CD8⁺ T cells around blood vessels and within brain tissue. These cells induce apoptosis of JC virus-infected oligodendrocytes through granzyme B, leading to demyelination and tissue damage [20]. It remains to be assessed whether the CD8+ T cells causing IRIS are truly HLA-restricted virus-specific T cells or are aspecific killers emerging during the early immune reconstitution phase. Indeed, relatively few patients treated with virus-specific T cell therapy experienced IRIS [11, 22].
To our knowledge, this is the first case describing PML-IRIS in LOCID patients. A potential trigger for its development could have been the immune-reconstitution induced by IVIG therapy, although data on this matter are lacking. In line with clinical practice guidelines primarily derived from the treatment of PML-IRIS in people with HIV, a course of steroid therapy was administered and proved successful, with no lesions documented in the follow-up MRI. An alternative approach could have involved targeting CCR5⁺ T cells, given their role in PML-IRIS. Maraviroc, a CCR5 antagonist, has been used to limit their migration into the CNS, though clinical outcomes have been inconsistent, and its benefit remains uncertain [1].
The favorable course of our patient may also be attributed to the clearance of the JCPyV in the cerebrospinal fluid. Existing evidence suggests a correlation between CSF viral load and improved prognosis in patients with PML [11, 23]. However, clinical outcomes after PML are highly variable, influenced by factors such as disease extent and timing of diagnosis. Data on long-term survivors remain limited, but many experience residual neurological deficits, with seizures reported in up to 44% of cases [1], as seen in our patient.
Although our follow-up extends to approximately two and a half years, it remains insufficient to fully assess long-term outcomes and reflects only one possible disease trajectory.
In conclusion, this case highlights the diagnostic and therapeutic challenges of managing PML in patients with latent immunosuppression, given the rarity of the condition and the limited evidence available to guide treatment decisions.
Author contributions
L.M.: Conceptualization, original draft writing, literature review. M.L.B., M.R.P.: Patient care, data analysis. P.C.: Laboratory investigation, writing. M.R.: Patient care, writing. G.L., P.B.: Writing, supervision. All authors reviewed the final version of the manuscript.
Conflicts of interest
The authors declare no relevant conflicts of interest related to this article.
Funding
This study received no external funding.
REFERENCES
- Cortese I, Reich DS, Nath A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat Rev Neurol. 2021; 17(1): 37-51.
- Jain V, Branstetter H, Savaram S, et al. Progressive multifocal leukoencephalopathy without overt immunosuppression. Medicine (Baltimore). 2023 Sep 29; 102(39): e35265.
- Berger JR, Aksamit AJ, Clifford DB, et al. PML diagnostic criteria: consensus statement from the AAN Neuroinfectious Disease Section. Neurology. 2013; 80(15): 1430-1438.
- Malphettes M, Gérard L, Carmagnat M, et al. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis. 2009; 49(9): 1329-1338.
- Kurmann R, Weisstanner C, Kardas P, et al. Progressive multifocal leukoencephalopathy in common variable immunodeficiency: mitigated course under mirtazapine and mefloquine. J Neurovirol. 2015; 21(6): 694-701.
- Du Pasquier RA, Kuroda MJ, Zheng Y, et al. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain. 2004; 127(Pt 9): 1970-1978.
- Ferenczy MW, Marshall LJ, Nelson CD, et al. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 2012; 25(3): 471-506.
- Lindner JM, Cornacchione V, Sathe A, et al. Human memory B cells harbor diverse cross-neutralizing antibodies against BK and JC polyomaviruses. Immunity. 2019; 50(3): 668-676.e5.
- L’Honneur AS, Pipoli Da Fonseca J, Cokelaer T, Rozenberg F. JC Polyomavirus whole genome sequencing at the single-molecule level reveals emerging neurotropic populations in progressive multifocal leukoencephalopathy. J Infect Dis. 2022; 226(7): 1151-1161.
- Lima MA, Bernal-Cano F, Clifford DB, Gandhi RT, Koralnik IJ. Clinical outcome of long-term survivors of progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 2010; 81(12): 1288-1291.
- Berzero G, Basso S, Stoppini L, et al. Adoptive transfer of JC virus-specific T lymphocytes for the treatment of progressive multifocal leukoencephalopathy. Ann Neurol. 2021; 89(4): 769-779.
- Pecoraro A, Crescenzi L, Granata F, Genovese A, Spadaro G. Immunoglobulin replacement therapy in primary and secondary antibody deficiency: the correct clinical approach. Int Immunopharmacol. 2017; 52: 136-142.
- Papakonstantinou I, Baraboutis IG, Karnesis L. Late onset combined immunodeficiency presenting with recurrent Pneumocystis jiroveci pneumonia. Case Rep Med. 2014; 2014: 801805.
- Lodha N, Shankar Meena D, Bhellum P, et al. Aspergillus terreus pulmonary infection in a patient with late-onset combined immunodeficiency: a case report with literature review. Ther Adv Infect Dis. 2024; 11: 20499361241265932.
- Paquin-Proulx D, Santos BA, Carvalho KI, et al. IVIg immune reconstitution treatment alleviates the state of persistent immune activation and suppressed CD4 T cell counts in CVID. PLoS One. 2013; 8(10): e75199.
- Kaveri SV, Maddur MS, Hegde P, Lacroix-Desmazes S, Bayry J. Intravenous immunoglobulins in immunodeficiencies: more than mere replacement therapy. Clin Exp Immunol. 2011; 164(Suppl. 2): 2-5.
- Cortese I, Muranski P, Enose-Akahata Y, et al. Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N Engl J Med. 2019; 380(17): 1597-1605.
- Sospedra M, Schippling S, Yousef S, et al. Treating progressive multifocal leukoencephalopathy with interleukin 7 and vaccination with JC virus capsid protein VP1. Clin Infect Dis. 2014; 59(11): 1588-1592.
- Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011; 11(5): 330-342.
- Bauer J, Gold R, Adams O, Lassmann H. Progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome (IRIS). Acta Neuropathol. 2015; 130(6): 751-764.
- Bernard-Valnet R, Moisset X, Maubeuge N, et al. CCR5 blockade in inflammatory PML and PML-IRIS associated with chronic inflammatory diseases‘ treatments. Neurol Neuroimmunol Neuroinflamm. 2021; 9(1): e1097.
- Cortese I, Beck ES, Al-Louzi O, et al. BK virus-specific T cells for immunotherapy of progressive multifocal leukoencephalopathy: an open-label, single-cohort pilot study. Lancet Neurol. 2021; 20(8): 639-652.
- Delbue S, Elia F, Carloni C, et al. JC virus load in cerebrospinal fluid and transcriptional control region rearrangements may predict the clinical course of progressive multifocal leukoencephalopathy. J Cell Physiol. 2012; 227(9): 3511-3517.